Genetic Map Details the Function of Almost Every Stem Cell Gene
Using Perturb-seq and CRISPR technology, researchers have developed a comprehensive reference atlas detailing how individual genes control human stem cell identity.
Genetic Map Details the Function of Almost Every Stem Cell Gene
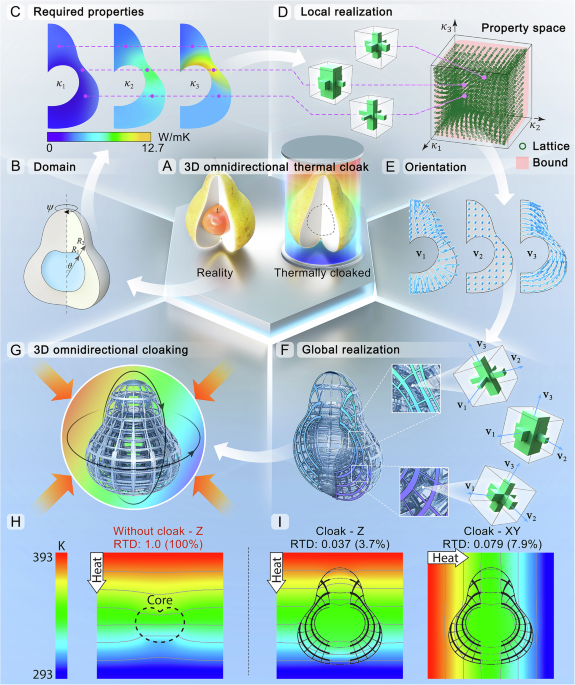
Bioengineers at the University of California San Diego have developed a genome-scale reference map that details how individual genes control the functions and identities of human stem cells. This open-access resource could help researchers build virtual cell models for complex diseases, as well as design patient-specific treatments for these diseases.
This is the first genome-scale map of gene function in human induced pluripotent stem cells. These are adult cells that have been reprogrammed back into an embryonic-like state and can turn into any type of cell in the body, such as muscle, heart, skin or bone. According to senior author Prashant Mali, professor in the Shu Chien-Gene Lay Department of Bioengineering at the UC San Diego Jacobs School of Engineering, the result is a reference atlas
that allows researchers to determine the impact of perturbing almost any gene on a stem cell’s behavior by measuring the effect on its whole transcriptome.
The Perturb-seq Methodology
To construct the map, the team used CRISPR technology to systematically switch off 11,692 expressed genes. The researchers then measured the resulting effects on cellular transcriptomes across more than 2.5 million single cells. This approach, known as Perturb-seq, uses CRISPR-Cas9 genome editing to introduce genetic changes into cells, and then uses single-cell RNA sequencing to capture information about the RNAs that are expressed resulting from a given genetic change, which control all aspects of how cells behave.
While Perturb-seq was first published in 2016 by a group including MIT professor Jonathan Weissman and professor Aviv Regev, it could only be used on small sets of genes and at great expense. A scalable version was later developed through a collaboration involving Joseph Replogle, an MD-PhD student in Weissman’s lab, and partners including 10x Genomics and Britt Adamson of Princeton University. This scaled-up method was documented in a 2020 Nature Biotechnology proof-of-concept paper.
The UCSD team used the data to group related genes and cellular components based on shared molecular traits and functions. This process isolated previously hidden metabolic and self-renewal genes and uncovered previously unrecognized cell regulators. Specifically, the researchers identified the gene DBR1 as the main regulator for RNA editing, specifically the conversion of adenosine to inosine.
Expanding the Functional Landscape
Parallel efforts at the Massachusetts Institute of Technology and the Whitehead Institute have also pushed beyond the 2003 completion of the Human Genome Project to map genotype-phenotype relationships. Professor Weissman described the data as a resource for discovery-based research
that allows scientists to screen the database without having to do any experiments.
Using human blood cancer cell lines as well as noncancerous cells derived from the retina, the MIT-affiliated team performed Perturb-seq across more than 2.5 million cells. Their analysis led to several biological discoveries:
- Protein Complexes: The researchers identified C7orf26 as a 15th component of the Integrator protein complex, which played a role in creating small nuclear RNAs.
- Chromosomal Instability: The team performed the first genome-wide screen for factors that are required for the correct segregation of DNA, identifying genes that cause aneuploidy—a condition known as cells to lose a chromosome or pick up an extra one.
- Mitochondrial Stress: The study explored how nuclear DNA (with around 1,000 related genes) and mitochondrial DNA (which carry 13 genes) coordinate and are regulated in different cellular conditions, especially when a cell is stressed. Replogle noted that one benefit of having a separate mitochondrial genome might be having localized or very specific genetic regulation in response to different stressors.
Future Applications and Access
The UCSD reference map is available at https://y-doctor.github.io/KOLF2.1J_Perturbation_Cell_Atlas/. Yesh Doctor, a bioengineering PhD student and study co-first author, stated the map works as a hypothesis engine
for identifying genes worth pursuing as targets to drive differentiation into cell states of interest.
Researchers hope to apply these single-cell CRISPR screens to a wider variety of cells and in vivo samples. While cost remains a major limitation, some genome-scale libraries have been sequenced using a lower-cost, ultra-high throughput sequencing platform developed by Ultima Genomics, generating results equivalent to those sequenced on Illumina instruments.